 Contents:
Contents:Computer modeling with the ESFF forcefield of human dihydrofolate reductase ternary complex with NADPH and piritrexim (PTX) inhibitor.
Contents:
Dihydrofolate Reductase (DHFR, EC 1.5.1.3) plays an important role in human physiology. The blocking of its enzymatic activity is a key element in treatment of many diseases, including cancer and AIDS related infections. Many useful drugs have been developed to date, however, issues of toxicity and drug resistance make it imperative that new inhibitors of DHFR be designed that have increased selectivity and lower toxicity. In this paper, computer modeling data are presented for the interactions of PTX bound to human DHFR NADPH ternary complex.
The commercially available ESFF forcefield from MSI was used to develop a computer model of the DHFR, PTX and NADPH complex. Water shell of 6 Å was included. This model has been tested against the X-ray structure and results of semiempirical calculations performed for simpler systems. The MD trajectories were generated to check the conformational freedom of the inhibitor. The binding interactions of PTX in the natural L22F mutation of hDHFR that results in methotrexate resistance, was also studied.
These results show that the ESFF forcefield, developed mainly for organometallic systems, may be effectively used in studies of protein structures.
Introduction.
Dihydrofolate reductase (DHFR) is the enzyme catalyzing the reduction of 7,8-dihydrofolate to 5,6,7,8-tetrahydrofolate, with NADPH being a coenzyme in this process [1]. Tetrahydrofolate is the source of the methyl groups in the synthesis of purines and pyrimidines, as well as methionine and histidine. The importance of DHFR in the folate cycle in living cell processes suggested this enzyme as a possible target for drug development against different tumors and infections of different origin. Extensive research on antifolates resulted in such drugs as the potent antibacterial trimethoprim (TMP) [2] or the antineoplastic agent methotrexate (MTX) [3]. Classical antifolates that have a benzyol glutamate moiety require specific transport system in mammalian cells, therefore have limited use for some therapies. The ability to penetrate the cell independently from the folate transport system is one of the ways to avoid the methotrexate resistance developing in the tumor. Piritrexim (PTX, 2,4-diamino-6-(2,5-dimethoxybenzyl)-5-methylpyrido[2,3d]- pyrimidine), was developed as a lipophilic antifolate [4] to overcome transport difficulties. Piritrexim is also a drug of interest for the treatment of the opportunistic infections in AIDS patients. Among different methotrexate-resistant variants of human DHFR, the ones with Leu22 substituted by larger amino acids such as the Leu22 to Phe variant, are stable and reveal decreased binding affinity for MTX, while their overall catalytic activity is similar to that of wild type human DHFR (wt hDHFR). The inhibition constants (Ki, nM) determined for PTX are 0.0077 and 0.659 for wt hDFR and L22F wariant, respectively [5]. These differences might be the subject for rational analysis based on the crystal structure determination or molecular modeling. Cooperativity between NADPH and TMP binding to bacterial DHFR was supported by the crystallographic data [6 and refs. cited therein]. Also the series of crystal structures of bacterial DHFR and its complexes revealed subdomain motion in E. coli DHFR during the catalytic cycle [7]. Such conformational changes might explain the cooperativity of substrate and coenzyme binding.
X-ray diffraction studies of the DHFR complexes have been the basis for the development of new antifolate drugs targeting specific organisms. However, computer simulations may also prove as a fast way of testing of the new molecules in the DHFR system. For example, molecular mechanics studies of PTX using YETI [8] and MM3 [9] forcefields proved useful in predicting the molecular geometry of PTX to be close to that observed in the PTX crystal structure. Moreover, Sutton and Cody used YETI [10] in a molecular modeling study to predict PTX binding interactions in chicken liver DHFR. Since that time, DHFR has been widely studied by computer simulations on both mechanical [11,12] and dynamical [13-18] levels. Much attention was also focused on the importance of electrostatics in the DHFR system [19,20]. C.S. Verma et al. conducted extensive molecular dynamics simulations of domain motions in L. casei DHFR complex with MTX, NADPH and 268 crystallographic water molecules [21]. More recently, a computational study of the affinity of 2,4-diaminopteridine hDHFR inhibitors was published by Aqvist and co-workers [22]. These authors used the GROMOS forcefield and a new, semi-empirical technique, LIE [23] to calculate absolute free energies of binding. Interestingly, L22Y and L22F mutants were addressed in this paper. However, no molecular dynamics simulations have been performed for human DHFR-PTX-NADPH ternary complexes.
One of the major obstacles in effective computer-aided drug design is a lack of reliable parameters for modeling of newly developed compounds. So far, several attempts have been made to establish more or less "automatic" procedures to generate computer models of any molecule. This philosophy was adopted, for example, in the UFF [24], Dreiding [25] and quite interesting ESFF formulations [26,27], covering almost all periodic table. Since DHFR is one of the most promising targets in drug design, it is a convenient system to validate (for doing massive calculations) the accuracy for the description of any ligand and the enzyme itself. We decided therefore to check the performance of the ESFF forcefield for such a system.
The aim of this study was to understand in greater detail the PTX binding mode in human DHFR. A secondary goal was to use the generic Extensible Systematic Forcefield (ESFF) available in the Insight 97.0 MSI software to study the dynamic behavior of the ligand in DHFR. Moreover, a comparison of theoretical models and crystal structures of DHFR-PTX-NADPH complex [28] and L22F DHFR-PTX-NADPH complex [5] has been performed. To our best knowledge, this is the first theoretical paper devoted to the PTX ligand and the important L22F mutant of human DHFR.
Methods
Quantum-chemical geometry optimization of PTX and charge distribution calculations were performed using the AM1 [29] method.
The ESFF forcefield (see for example [27]), as implemented in InsightII v. 97.0 package [30] was chosen for its ease in parametization of all atom types automatically (up to radon), and its ability to recalculate on-the-fly the partial charges based on the local environment of the atoms. It is a diagonal forcefield, with Morse bond potentials and based on density functional theory calculations of atomic electronegativity and hardness. The charges are determined in a rather complex way, by minimizing the electrostatic energy under the constraints that the sum of charges is equal to the net charge of the molecule. The ESFF uses the 6-9 potential for the van der Waals interactions. In this forcefield, van der Waals parameters are affected by the charge of the atom. An important part of modeling is correctly setting up all the bond orders in the system under study.
The ESFF forcefield has been used by others for simulations of organometallic compounds [26,27], but it may be also applied to typical biomolecules. The usefulness of the generic ESFF forcefield for simulation of the protein system might be important for those involved in the research of biological systems, and not willing to develop elaborate parameters for exotic molecules of interest, especially those having organometallic groups.
The X-ray crystal structure of wt DHFR-PTX-NADPH complex [28] refined to 2.3 Å was used as a starting point for further modeling. All 100 crystallographic water molecules were retained in the model. Hydrogen atoms were added using the "Biopolymer" module of the InsightII package. The same module was used to substitute the leucine in position 22 by phenylalanine. The "soak" option of "Biopolymer" was used to generate a 6 Å layer of water molecules (1693 water molecules) resulting in 7625 atom model of solvated complex (see Fig. 1). The PTX ligand was protonated at N1 atom (net charge +1) and NADPH had net charge of -3. The DHFR amino acids interacting with both ligands (Glu30, Lys54, Lys55, Arg77) were charged giving the zero total charge of the model complex. The group based method was used in the nonbonded interactions energy evaluation. Both wt and L22F hDHFR models were subjected to 400 steps of conjugate gradient method (Polak-Ribiere) minimization (module "Discover3" of InsightII v 97.0), proceeded by 20 steps of water shell optimization with the protein part held fixed, leading to enrgy gradients of 10.2 and 9.3 kcal/mole/Å, respectively. At this minimization stage, a 30 Å nonbonded interactions cutoff with 1 Å wide buffer switching region was used. Such a large value of the cutoff at this stage was deliberately used to encompass a large NADPH group and to include long-range electrostatic effects. The ESFF partially minimized structures of both wt hDHFR and L22F mutant were compared with the X-ray structures.
Fig. 1. [65k] The DHFR-NADPH-PTX system constructed by adding the 6 Å layer of water molecules to the hydrated protein complex as determined by X-ray crystallography [28]
For molecular dynamics (MD) simulations, a large cutoff of 30 Å is not practical. In order to reduce computational time we lowered this value to 15 Å, divided the PTX ligand and NADPH coenzyme into small charge groups, and subjected the structure to a further 20 steps of minimization before the heat stage and MD runs. A dielectric constant of 1 was used through all computations. All bond lengths were kept constant using RATTLE algorithm [31]. The simulation timestep was 1 fs. The initial temperature was 10 K. During the dynamics simulations it was kept constant at 298 K by a velocity scaling. Since we intended primarily exploratory calculations, the heating and equilibration phase were severely reduced (<1000 steps). The data were collected through 10 ps, each 0.1 ps, and analyzed using InsightII 97.0 and PROCHECK [32] tools. Calculations were performed on the SGI O2 R5000 workstation.
Results and Discussion
PTX geometry, conformation and charges
It is known that PTX is a flexible molecule due to the presence of the methylene bridge [8,9]. Recent MM3 computations of potential energy surface for PTX showed that the barriers between distinct conformational minima do not exceed 2 kcal/mol [9]. This flexibility manifests itself in our results as well. The conformation of PTX found in the small molecule crystal structure [8], the wt hDHFR complex [28], AM1 minimized and ESFF minimized structures differ substantially in torsion angles around the bridging methylene group. The comparison of PTXH+ cation from AM1 and the structure from protein crystal is shown on Fig. 2.
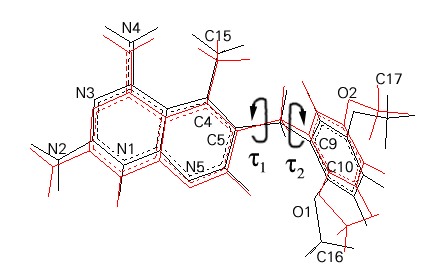 Fig. 2. Comparison of AM1 minimized (red) and crystal (black) PTXH+ cation geometry. The arrows indicate the torsion angles t
1 (C4-C5-C8-C9) and t
2 (C5-C8-C9-C10)
Fig. 2. Comparison of AM1 minimized (red) and crystal (black) PTXH+ cation geometry. The arrows indicate the torsion angles t
1 (C4-C5-C8-C9) and t
2 (C5-C8-C9-C10)The PTX geometry from the protein crystal was used to construct the PTXH+ monocation protonated at N1 atom of the pteridine ring. Such an entity is expected to exist in the DHFR binding site and to form the double salt bridge to the enzyme Glu30 side chain. This model was a starting point for the ESFF energy minimization, as well. The superposition of the ESFF minimized, protein crystal [28] and PTX crystal [8] structures is shown in Fig. 3. The bond distances and bond angles of these monocations are similar, with differences (up to 5o) only observed in the vicinity of the protonation site. This geometry perturbation is understandable since in the PTX crystal structure [8] the molecule is not protonated. However, the effect of semi-empirical and ESFF minimization is visible in the bridge conformation. While the t 1 (C4-C5-C8-C9) and t 2 (C5-C8-C9-C10) torsion angles of -76.3o, 146.0o, respectively, have been found in the crystal structure [8], the corresponding values after AM1 and ESFF minimization were 109.9o, -88.9o and -178.9o, -78.5o, respectively. The comparison of the conformation of PTX as found in the minimized wt and L22F mutant complexes (see next paragraph) is presented in Table 1.
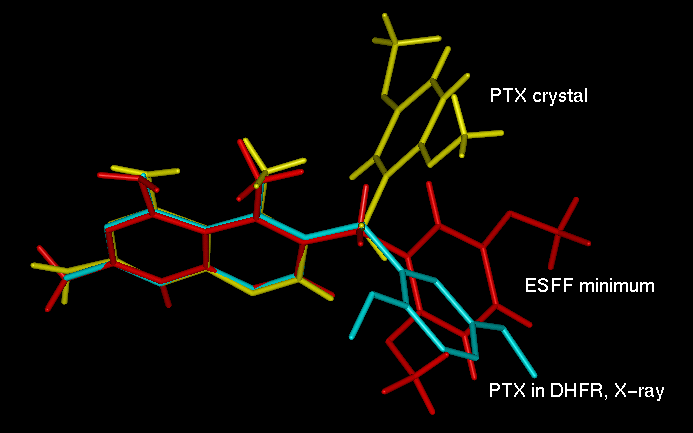
Fig. 3. Superposition of PTX from small molecular crystal structure [5] (yellow), wt hDHFR complex [28] (cyan) and ESFF minimized isolated inhibitor molecule (red).
Table 1. Conformation of the PTX ligand described by selected torsion angles, as found in the crystal structure of wt hDHFR ternary complex, the minimized structure and the L22F ternary complex built with the MSI software and minimized.
|
Selected torsion angles |
Crystal wt DHFR complex |
wtDHFR complex minimized |
L22F complex minimized |
|
C4-C5-C8-C9 |
145.1 |
119.0 |
151.8 |
|
C5-C8-C9-C10 |
-90.8 |
-83.9 |
-105.0 |
|
C9-C10-O1-C16 |
146.2 |
139.2 |
103.3 |
In all structures the orientation of methoxy groups planes have been found close to the phenyl ring plane, with the dihedral angle not exceeding 45o. Interestingly, we found that the two methoxy groups have systematically different orientations with respect to the phenyl plane: while the group in the meta position is almost coplanar, the group in the ortho position shows larger values of the dihedral angle, namely: 13.8o (small molecule), 19.7o (AM1), 33.8o (protein) and 45.2o (ESFF). The above results of the ESFF parametrization of the PTXH+ system are reasonable and are consistent with both crystallographic and quantum chemical results. The ESFF deviations from crystal data of 5o in the bond angles or smaller than 0.09 Å in the bond length should not be a decisive factor for the successful simulation of the large system as this 20 kDa protein complex.
It was not our goal to optimize charges for this enzyme complex, and PTX in particular. However, the results presented in Fig. 4 give an example of the relationship between ESFF generated charges and those commonly used for AM1 partial charges. One can see that qualitatively both sets are similar. In principle, the protein environment might lead to strong polarization of the ligand [19]. To check this effect for the PTX ligand embedded in the DHFR binding site, we calculated a charge distribution in a supermolecule consisting of PTX and close lying residues Ile7, Glu30, Val115, WAT252 molecule and a nicotinamide part of the NADPH (see Fig. 5). The protein complex geometry and the AM1 method were used. These results show, that the effect of these selected amino acids on the PTX charge distribution, at least on the AM1 method level, is negligible.
Fig. 4. [23k]ESFF and AM1 partial charges for PTXH+ monocation.
Fig. 5. [28k]The AM1 partial charges for the assembly of PTX (cyan) with Glu30, Ile7, Val115 (green), Wat252 (red) and nicotinamide (yellow) in the geometry found in the crystal structure of wt hDHFR complex [28].
Minimization of hDHFR ternary complexes
The wt DHFR complex was used to model the methotrexate-resistant L22F mutant. Both the systems were minimized using the ESFF forcefield. The results of the minimization were compared to the crystal structure of the hDHFR-NADPH-PTX complex [28]. The ESFF minimization resulted in the structure similar to the starting one. The rms differences calculated for the Ca atoms of the enzyme, and all atoms of NADPH and PTX, are 0.62 Å, 0.66 Å and 0.58 Å, respectively. The overall conformation of the polypeptide chain of DHFR was maintained during the minimization. The PTX monocation was re-positioned to fit the tilt of the MTXT pteridine ring system, while maintaining the salt bridges to Glu30. The slight rotation of the methoxy group was found which resulted in weaker interactions to ribose ring of NADPH. Slight changes in the pyrophosphate conformation and ribose moieties were also found, but the final position of the nicotinamide moiety was similar to that observed in the crystal structure of hDHFR-NADPH-PTX ternary complex (Fig. 6). No significant change in the side chain conformation of the amino acids comprising the active site was found (Fig. 7). The major changes in the vicinity of the binding cleft are observed for Pro61 and Glu62, which are not well defined in the crystal structure. The differences in Ca positions are 1 Å for both residues. For the rest of the residues constituting the PTX binding site, these differences are 0.2-0.3 Å. These changes reflect the relaxation of the strain resulting from the compromise between the electron density constraints and the energetics of the CHARMM forcefield in X-PLOR [33] during refinement of the crystal structure. The rms differences for Ca atoms indicate, however, that the conformational changes are small, and the overall polypeptide conformation is well maintained by the ESFF based minimizer.
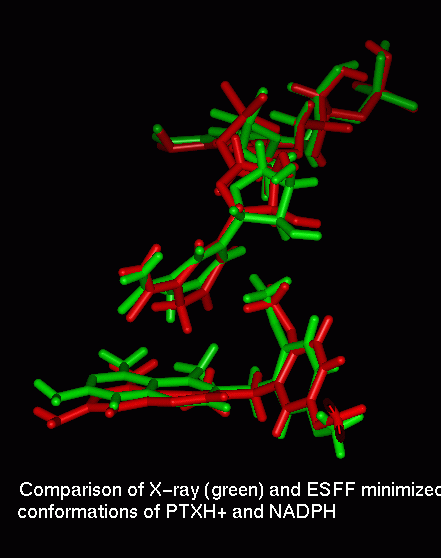
Fig. 6. Superposition of PTX and NADPH from the crystal structure [28] and ESFF minimal structure.
The PROCHECK program [32] was used to monitor the conformation of the main chain and side chains, and to assess the overall quality of the minimized polypeptide geometry. The most striking result of the 200 cycles of energy minimization was the lost of planarity for the peptide bonds (Table 2). The starting crystal structure was refined with X-PLOR [32] and CHARMM forcefield to the resolution of 2.3 Å. The comparison of the polypeptide chain geometry revealed that the ESFF minimization resulted in the less tight restraints on the phi/psi angles. In the crystal structure 91.8% of amino acids have been found in the most favored regions of the Ramachandran plot, and remaining 8.2% in the allowed regions. In the structure minimized with ESFF, the percentage of residues in the most favored, allowed and generously allowed regions were 84.3, 14.5 and 1.3%, respectively. The relative loss of the geometry quality for the main chain was not dramatic, and the visual inspection of the structure has not revealed any unfolding of the polypeptide chain, even in the terminal regions of the protein. The global behavior of the system during minimization was quite satisfactory, with the Ramachandran region distribution, bad contact number, configuration of Ca atoms and H-bonding inside the range of parameters for the crystal structures determined to 2.3 Å (resolution for the starting crystal structure). The only parameter worse than the average for the crystal structures, and significantly worse than that of the starting structure, was the omega torsion angle describing the planarity of the peptide bond. The e.s.d for this value in the crystal structures is 6.0o+-3.0o. In the starting structure it was 2.2o, while for the minimized system it was 9.2o.
Table 2. The main chain parameters for the main chain atoms for the crystal structure and ESFF minimized ternary complexes of wt hDHFR and its L22F variant.
|
Stereochemical Parameter |
wt hDHFR-NADPH-PTX Crystal structure |
Typical value for structures at 2.3 Å resolution |
wt hDHFR-NADPH-PTX Minimized - ESFF |
L22F hDHFR-NADPH-PTX Minimized - ESFF |
|
% Most favored |
91.8 |
82.6+-10.0 |
84.3 |
80.5 |
|
Omega angle st.dev. |
2.2 |
6.0+-3.0 |
9.2 |
9.1 |
|
Bad contacts/100res. |
1.1 |
5.2+-10.0 |
0.5 |
0.5 |
|
Zeta angle st.dev. |
1.8 |
3.1+-1.6 |
2.0 |
1.9 |
|
H-bond E st.dev. |
0.8 |
0.8+-0.2 |
0.8 |
0.7 |
The side chain conformation for all the residues, monitored in terms of Chi1/Chi2 pairs of torsion angles, showed some changes due to the ESFF minimization, with the insignificant improvement after minimization. The Chi1 distribution in the minimized structure was better than average and for all categories slightly closer to the standard value (+-60o, 180o) than found in the starting crystal structure (Table 3). The similar quality of the structure may be connected to the use of the energy terms in both crystal structure refinement and the minimization. The constraints originated in the experimentally measured structure factors, with all the experimental errors, gave 6 out of 186 side chains distant from the ideal values by more than 2.5 standard deviations. The lack of these constraints during ESFF energy minimization resulted in a decrease of this number to 4, which might correspond to the better intermolecular interaction network possible but not observed in the crystal structure. Also, the additional layer of water added to the system might change the pattern of polar interactions resulting in the different energy minimum.
Table 3. The standard deviations of the side chain angles for the wt hDHFR complex crystal structure and ESFF minimized ternary complexes of wt hDHFR and its L22F variant.
|
Stereochemical Parameter standard deviations |
wt hDHFR-NADPH-PTX Crystal structure |
Typical value for structures at 2.3 Å resolution |
wt hDHFR-NADPH-PTX Minimized in ESFF |
L22F hDHFR-NADPH-PTX Minimized in ESFF |
|
Chi1 gauche(-) |
17.2 |
19.0+-6.5 |
8.2 |
7.6 |
|
Chi1 trans |
14.0 |
19.7+-5.3 |
16.0 |
15.5 |
|
Chi1 gauche(+) |
13.8 |
18.3+-4.9 |
9.8 |
10.8 |
|
Chi1 pooled. |
14.7 |
18.9+-4.8 |
13.0 |
13.0 |
|
Chi2 trans |
14.2 |
20.9+-5.0 |
10.5 |
11.6 |
The steric and energetic perturbation was introduced to the minimized structure by modeling of the L22F mutant. The Leu22 side chain is positioned inside the substrate binding site of DHFR enzyme. The replacement of Leu by a larger side chain of Phe caused additional repulsive interactions with PTX in the narrow part of the binding cleft. The resulting system was subjected to additional 200 cycles of minimization. The distribution between the most favored, allowed and generously allowed regions of Ramachandran plot was further distorted, with the corresponding populations of 80.5, 17.6 and 1.9%. Again, no dramatic collisions or protein unfolding was observed.
The above analysis shows that the moderate ESFF minimization of the DHFR protein ternary complex leads to a realistic structure. The planarity of the peptide bonds may be, however, of some concern in certain ESFF protein models.
The ligand interactions
The PTX ligand is positioned differently in both the minimized wt DHFR complex structure, when compared to the starting crystal structure. The pteridine ring is shifted by 0.5 Å along the ring plane towards the Glu30 carboxylic group and tilted around the central bridging bond by 14o (change in dihedral angle), see Fig. 6. Simultaneously, the minimization resulted in the change of the bridge C4-C5-C8-C9 torsion angle from 145.1 to 119.0o and C5-C8-C9-C10 from -91.0 to -84.0o. These changes result from the narrowing of the binding cleft near the dimethoxyphenyl moiety of PTX, since the Pro26 Ca to Pro61 Ca distance has decreased from 8.78 to 7.45 Å.
Conformation of PTXH+ ligand is slightly different between two minimized complex structures due to the steric contribution of Phe22 (Table 1). In the new conformation the piritrexim ligand fits better to the space available between the side chains of Ile60, Pro61, Phe31 and Phe22. The ligand conformation is also changed during the energy minimization. These differences might reflect the absence of the constraints from the crystallographic data on the position of the ligand in the catalytic site of the enzyme.
Minimization also changes the conformation of NADPH coenzyme. While the adenosine and nucleotide moieties are almost not changed, with only slight adjustments of the pyrophosphate conformation observed (see Fig. 6). The reorientation of the sugar ring between the pyrophosphate and the nicotinamide moiety compensate for these changes, and the latter occupies the same space near PTX as before the minimization. The polar interactions of NADPH with surrounding amino acids are preserved during the minimization.
The conformation of Phe31 is particularly interesting since it is positioned in the center of the folate/inhibitor binding site and has been shown flexible in other crystal structures. The conformation of Phe31 found in the crystal structure of wt DHFR ternary complex (180o, 58o) remained unchanged during the ESFF minimization (-173o, 53 ) but changed significantly for the constructed and minimized L22F complex structure (-170o, 17o). The close examination of all three structures suggested, that the observed change is due to the repulsive interactions between the large Phe22 side chain and PTX, resulting in this unusual conformation of the Phe31 side chain. Since in the X-ray structure of the L22F variant the conformation of this residue is (-178.5o, 52.4o), it seems that a more elaborate search for minimum energy structure, including simulated annealing, should be performed in the case of computer generated mutants, to prevent making wrong conclusions regarding effects of mutations.
There is a conserved water molecule found in the substrate binding site in all the crystal structures of DHFR complexes. This water molecule (see Fig. 5) in the wt hDHFR-NADPH-PTX complex is involved in the interactions to Trp24 NE1, Glu30 OE2 and PTX N5 (3.33, 2.92 and 3.88 Å, respectively). During the energy minimization this water molecule is slightly shifted, with the corresponding distances being 2.81, 2.86 and 3.53 Å, respectively, the above changes reflecting not only the forcefield effects but also the repositioning of the PTX ligand during minimization. In the L22F minimized structure the water interactions to W24 and E30 are maintained. The PTX is tilted towards the opposite side of the binding cleft, therefore loosing the contact to the observed position of the conserved water. The corresponding distances are 3.00, 2.89 and 4.30 Å, respectively.
The authors of the ESFF forcefield focused primarily on reliable structure predictions, so the interaction energies calculated using this universal approach do not necessarily have to be highly realistic [30]. Despite this caveat, we tried to estimate qualitatively the interactions in which the DHFR ligands are involved. The comparisons with the experimental kinetic or activity data may be done only on the basis of free energy calculations. However, the present analysis, in our opinion, may indicate interesting qualitative differences between the binding to the DHFR variants.
The interaction energy between the PTX monocation and other parts of the system was calculated for the wt and L22F DHFR complexes (Table 4). The major contribution of the PTX binding is due to the double salt bridge formed to Glu30 side chain (Fig. 7). Also the electrostatic part of the Ile7 and Val115 interactions to the amino group of PTX strengthen the ligand binding in the active site of DHFR. The significant contribution to the inhibitor binding is the coulomb interaction with the nicotinamide-ribose moiety of the coenzyme. The interesting result is that in the reported position of PTX, different from that of MTXT [34], the interactions with Leu22, Phe31 and Phe34 are negligible.
Table 4. The piritrexim (PTX) intermolecular interaction energy (kcal/mol) calculated for wt hDHFR and L22F hDHFR ternary complexes using ESFF forcefield in Discover3. The calculated energy is partitioned between the selected amino acids in the binding site, whole protein, NADPH and water. Nonbonded cutoff of 15.0 Å was used.
|
PTX interactions |
wt hDHFR-NADPH-PTX (Leu-22) |
L22F hDHFR-NADPH-PTX |
||||
|
Coulomb |
vdW |
Total |
Coulomb |
vdW |
Total |
|
|
Ile-7 |
-2.50 |
-0.40 |
-2.90 |
-2.50 |
-0.65 |
-3.14 |
|
Leu/Phe-22 |
1.18 |
-2.15 |
-0.97 |
0.77 |
-2.31 |
-1.54 |
|
Glu-30 |
-76.62 |
5.51 |
-71.11 |
-66.56 |
4.16 |
-62.40 |
|
Phe-31 |
0.22 |
-1.82 |
-1.60 |
-0.97 |
-2.28 |
-3.26 |
|
Phe-34 |
2.61 |
-2.31 |
0.30 |
2.29 |
-3.03 |
-0.7 |
|
Val-115 |
-4.29 |
-0.32 |
-4.62 |
-4.40 |
-0.59 |
-4.980 |
|
Protein total |
-36.64 |
-15.52 |
-52.15 |
-37.06 |
-15.80 |
-52.86 |
|
NADPH total |
-35.92 |
-2.72 |
-38.64 |
-34.99 |
-2.24 |
-37.23 |
|
Water total |
-0.37 |
-0.21 |
-0.54 |
0.98 |
-0.33 |
0.65 |
|
Total |
-72.90 |
-18.44 |
-91.34 |
-76.08 |
-18.37 |
-89.45 |
Thus the major component of the calculated PTX-protein interaction energies in wt and L22F variant is electrostatic interaction. Low values of PTX interactions with water are probably related to the fact that the ligand is buried a hydrophobic cleft.
The molecular dynamics simulations
Both wt and L22F structures were subjected to 10 ps MD simulation (further simulations are in progress). The short trajectories do not deliver sufficient sampling of the whole phase space, however, are good enough to grasp major features of the tested forcefield. The mean square deviation (msd) from the starting structure was calculated for the whole structure, the protein atoms, NADPH and PTX separately (all atoms). The values for NADPH and PTX reached 1.56 Å2 and 4.58 Å2 for wt and 3.57 Å2 and 3.62 Å2 for L22F, respectively, reflecting the stable positions of both molecules with some degree of conformational flexibility. The msd values for the enzyme atoms are 2.65 Å2 and 2.64 Å2 for wt and L22F, respectively.
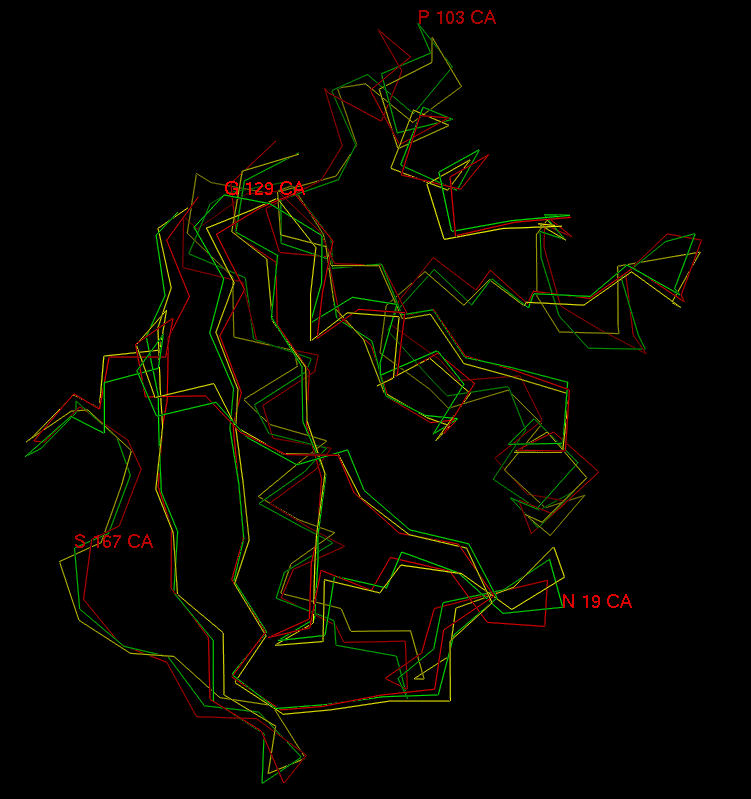
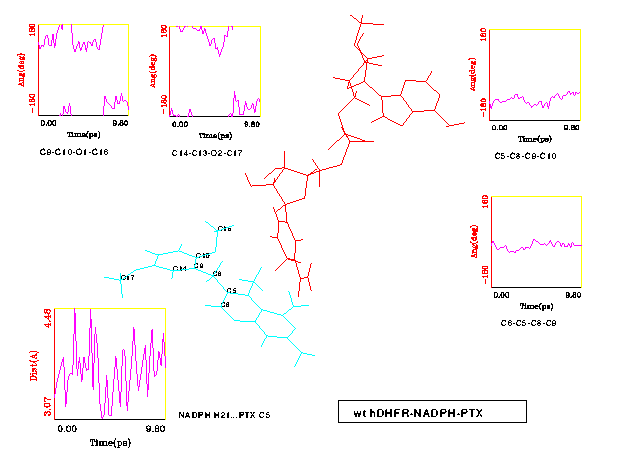
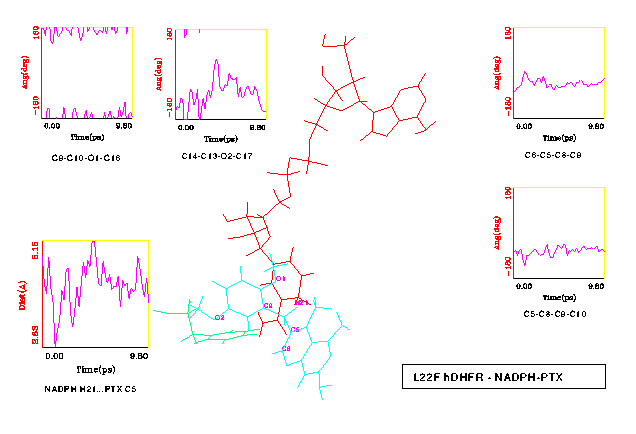
Visual inspection of both wt and L22F trajectories using computer graphics indicate that the simulated complexes are stable on this short timescale. The protein backbone does not change substantially - the rms differences for all backbone atoms between wt crystal structure and 10 ps snapshots of both wt and L22F is 1 Å (see Fig. 8). Water shell undergoes intensive rearrangement, but despite of a lack of any constraints only one water molecule diffused away from the system. In Fig. 9 and Fig. 10 the selected time-series for wt and L22F variant are presented. We focused here on the PTX structure time evolution and changes in NADPH-PTX distance. The critical distance for the enzymatic function of NADPH is that involved with hydride transfer from NADPH and the folate substrate. We have found that pteridine C5 - H21 (nicotinamide) distance ranges from 3.07 to 4.48 Å for wt (Fig. 9) and from 2.83 Å to 5.15 Å in L22F (Fig. 10). This distance fluctuates around 3.8 Å in wt and 4.2 in L22F variant. Our data indicate that fluctuations in the critical distance in native protein are large, close to 2 Å. Moreover, we found that the PTX inhibitor remains close to the catalytic part of the hydride donor (coenzyme). The ESFF forcefield does not destroy this important feature of the system studied.
The most interesting differences were observed for the flexibility of the two methoxy groups of PTX. In the wt DHFR complex these group change conformation in a similar manner (see Fig. 9). On the other hand, the reorientation of the inhibitor in the L22F complex resulted in the 2'-methoxy group positioned slightly closer to the NADPH sugar ring, therefore restricting the fluctuations of this group compared to the 5'-methoxy (see Fig. 10).
The rotational freedom on the methylene bridge of the PTX inhibitor is restricted in the protein cleft. Indeed, the maximum observed deviation from minimized structure values in torsional degrees of freedom was 70o (Fig. 9). One may expect that L22F mutation should restrict conformational transitions in PTX in comparison with wt DHFR. Just the reverse trend is observed in MD results: fluctuations of the t 1 and t 2 angles are larger in L22F mutant than in the wt model. The cleft in DHFR is thus large enough that there is no unique way of PTX binding in this system. We observe that although PTX is being held by a double salt bridge to a flexible Glu30 side chain, it can still probe various regions of the binding pocket. This statement is illustrated in Fig. 11, where distinct orientations of the PTX dimethoxyphenyl moiety in representative time intervals are shown. One can see that this part of PTX is rather "vigorous" while the Glu30 bound pteridine part changes its position to a much smaller extent.
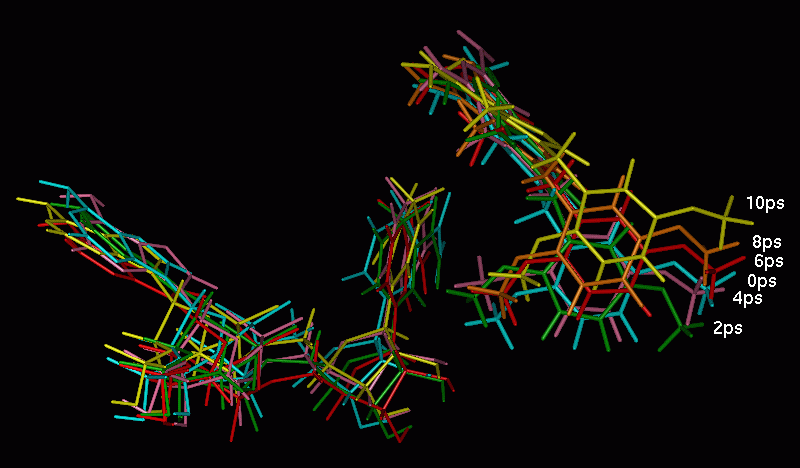
Fig. 11. Fluctuations of the PTX and NADPH positions during the MD simulation, shown at the selected snapshots for wt DHFR trajectory.
At the same time NADPH ion interacts strongly with the DHFR. On our short 10 ps timescale we observe substantial "breathing" motions of the coenzyme, which results in about 2 Å fluctuations in the adenosine-nicotinamide distance. As shown in Fig. 12, the overall position of the NADPH does not change much.
Fig. 12. [46k] The "breathing" motion of NADPH during the MD simulation of wt DHFR complex.
In general, also in dynamical part of the DHFR complexes modeling the ESFF forcefield performs rather well. One should note, however, that different scheme for charging protein residues or smaller cutoffs in nonbonded interaction may change this optimistic picture.
Conclusions
The ESFF forcefield gives the good quality results for the geometry of PTX ligand, and therefore might be used as a routine approach to the modeling studies of new ligands of interest.
The ESFF proved also to be useful for the simulations of the protein complex system, both starting from the crystal structure and from the model built based on the known crystal structure. Special caution should be devoted to the treatment of the polypeptide omega angle (Ca -C-N-Ca ) which tends do loose its planarity during minimization or MD simulation. It is worth to mention that during the minimization, the conformation of the polypeptide chain of our alpha/beta protein did not change significantly, even in the absence of the constraints from the experimental electron density, when compared to the crystal structure. Also the distribution of amino acids on the Ramachandran plot between the three allowed sections is encouraging for the future use of ESFF for the routine simulations of the similar proteins. Moreover, the minimized structures reveal that the average conformation of the side chains is even better than average for all crystal structures in PDB determined at the resolution of 2.3 Å corresponding to the starting structure.
The orientation of the NADPH ligand in the model complex is similar to that observed in the crystal structure. Despite some relaxation, due to the nature of the forcefield potentials, all important ligand-protein interactions are preserved during the energy minimization. Qualitative trends in the hierarchy of interaction energy of the PTX ligand were successfully reproduced. Using the ESFF forcefield the strongest interactions of PTX are predicted for the Glu30 and NADPH coenzyme.
In MD simulations some variation of the PTX conformation and orientation was observed, but the overall position of the inhibitor remains in good agreement with that found in the crystal structure. The difference found in the L22F complex reflects the change in the binding site geometry due to the larger volume of the F22 side chain, and is consistent with the reported crystal structure of L22F ternary complex. Also the MD simulation reveals that the fluctuations observed for NADPH are not very large. The PTX ring system remained bound to Glu30 carboxylic group in both wt and L22F variant, and the major fluctuations corresponded to a flexible conformation of the methylene bridge of the inhibitor. The distance of the C5 pteridine atom to the H21 nicotinamide reducing agent is similar in wt and L22F on 10 ps time scale studied here. This capability of PTX to adopt an orientation in L22F which is similar to that observed in wt explains why, in contrast to methotrexate, PTX drug is a good inhibitor of L22F variant as well.
The ESFF forcefield produced a reasonable estimation of the ligand binding interactions, confirming the major contribution from the Glu30 side chain. Another interaction dominating the PTX binding was that with NADPH. This seems to be consistent with the observation of cooperativity in binding of antifolate inhibitors and NADPH to DHFR.
The results of these short, 10 ps simulations seem to be quite promising. They prompted us to continue modeling of the DHFR systems using the ESFF forcefield. More work oriented on the ESFF validation for protein systems is clearly required. We hope that the universal character of ESFF approach will help to develop new, selective inhibitors of the DHFR enzyme.
Acknowledgments
This work was supported in part by KBN 6/P04A/032/11 (AW, WN), KBN 6/P04A/066/14 (WN), and FIRCA Fogarty R03 TW 00789-02 grants (VC, AW).
Computational results obtained using software programs from Molecular Simulations Inc. - dynamics calculations were done with Discover program, using the ESFF forcefield, graphical displays were printed out from the InsightII molecular modeling system. Authors thank KBN (Warsaw, Poland) and grant UMK (WN) for partial support of the nation wide license of MSI software.
1. Blakley, R.L. (1995). Advances in Enzymology and Related Areas of Molecular Biology, Vol. 70, pp. 23-102, John Wiley & Sons, Inc.
2. Hitchings, G.H. & Roth, B. (1980). Enzyme Inhibitors as Drugs (Sandler, M. ed.) pp. 263-280, Macmillan.
3. Duch, D.S., Edelstein, M.P., Nichol, C.A. (1982). Cancer Res. 42, 3987.
4. Grivsky, E.M., Lee, S., Sigel, C.W. & Nichol, C.A. (1980). J. Med. Chem. 23, 327.
5. Lewis, W.S., Cody, V., Galitsky, N., Luft, J.R., Pangborn, W., Chunduru, S.K., Spencer, H.T., Appleman, J.R. & Blakley, R.L. (1995). J. Biol. Chem. 270, 5057- 5064.
6. Champness, J.N., Stammers, D.K. & Beddel, C.R. (1986). FEBS Letters, 199, 61-67.
7. Sawaya, M.R. & Kraut, J. (1997). Biochemistry, 36, 586-603.
8. Sutton, P.A. & Cody, V. (1988). J. Am. Chem. Soc. 110, 6219-6224.
9. Fleisher, R., Wiese, M., Troschultz, R. & Zink, M. (1997). J. Mol. Model. 3, 338-346.
10. Vedani, A. (1988). J. Comput. Chem., 9, 269.
11. Roberts, V.A., Dauber-Osguthorpe, P., Osguthorpe, D.J., Levin, E. & Hagler, A.T. (1986). Isr. J. Chem. 27, 198-210.
12. Dauber-Osugthorpe, P., Roberts, V.A., Osguthorpe, D.J., Wolff, J., Genest, M. & Hagler, A.T. (1988). Proteins: Struct. Funct. Genet. 4, 31-47.
13. Sing, U.C. & Benkovic, S.J. (1988). Proc. Natl. Acad. Sci. USA, 85, 9519-9523.
14. Fleishman, S.H. & Brooks, C.L. III (1990). Proteins: Struct. Funct. Genet. 7, 52-61.
15. McDonald, J.J & Brooks, C.L. (1992). J. Amer. Chem. Soc., 114, 3062-3072.
16. Gerber, P.R., Mark, A.E. & Gunsteren, W.F. (1993). J. Comput. Aid. Mol. Des. 7, 305-323.
17. Cummins, P.L. & Gready, J.E. (1993). J. Comput. Aid. Mol. Des. 7, 535-555.
18. Leach, A.R. & Klein, T.E. (1995). J. Comp. Chem. 16, 1378-1393.
19. Bajorath, J., Kraut, J., Li, Z., Kitson, D.H. & Hagler, A.T. (1991). Proc. Natl. Acad. Sci. USA 88, 6423-6426.
20. Cannon, W.R., Garrison, B.J. & Benkovic, S.J. (1991). J. Amer. Chem. Soc., 119, 2386-2395.
21. Verma, C.S., Caves, L.S.D., Hubbard, R.E. & Roberts, G.C.K. (1997). J. Mol. Biol. 266, 776-796.
22. Marelius, J., Graffner-Nordberg, M., Hansson, T., Hallberg, A. & Aqvist, J. (1998). J. Comput. Aid. Mol. Des. 12, 119-131.
23. Aqvist, J., Medina, C. & Samuelsson, J.E. (1994). Protein Eng. 7, 384.
24. Rappe, A.K., Casewit, C.J., Colwell, K.S, Goddard, W.A. & Skiff, W.M. (1992). J. Amer. Chem. Soc., 114, 10024.
25. Mayo, S.L., Olafson, B.D. & Goddard, W.A (1990). J. Phys. Chem. 94, 8897-8909.
26. Barlow, S., Rohl, A.L. & O'Hare, D. (1996). Chem. Comm. 257-260.
27. Barlow, S., Rohl, A.L. Shi, S.G., Freeman, C.M. & O'Hare, D. (1996). J. Amer. Chem. Soc. 118, 7578-7592.
28. Cody, V., Wojtczak, A., Kalman, T.I., Freisheim, J.H. & Blakley, R.L., (1993). In: Chemistry and Biology of Pteridines and Folates, Ayling, J.E., Nair, M.G. & Baugh, C.M. (eds), Plenum Press, pp 481-486.
29. Dewar, M.S.J, Zoebish, E.G., Healy, E.F. & Stewart, J.J.P (1985). J. Amer. Chem. Soc., 107, 3902.
30. 'Forcefield-Based Simulations', April 1997, San Diego: Molecular Simulations Inc. 1997.
31. Andersen, H.C. (1983). J.Comp. Physics, 52, 24-34.
32. Laskowski, R.A., MacArthur, M.W., Moss, D.S. & Thornton, J.M. (1993). J. Appl. Cryst. 26, 283-291.
33. Brunger. A.T. (1992). X-PLOR Version 3.1: A System for X-ray Crystallography and NMR, Yale University Press, New Haven, CT, USA.
34. Cody, V., Luft, J. R., Ciszak, E., Kalman, T.I. & Freisheim, J.H. (1992). Anticancer Drug Design 7, 483-491.
![Fig. 1. [65k] The DHFR-NADPH-PTX system constructed by adding the 6 Å layer of water molecules to the hydrated protein complex as determined by X-ray crystallography [28]](fig1_p46.gif){kind=link}
![Fig. 4. [23k]ESFF and AM1 partial charges for PTXH+ monocation.](fig4_p46.gif){kind=link}
![Fig. 5. [28k]The AM1 partial charges for the assembly of PTX (cyan) with Glu30, Ile7, Val115 (green), Wat252 (red) and nicotinamide (yellow) in the geometry found in the crystal structure of wt hDHFR complex [28].](fig5_p46.gif){kind=link}
![Fig. 7. [39k] The protein binding interactions in the crystal structure (green) and ESFF minimal structure (red) of wt DHFR complex. (The question marks on the figure indicate that one letter code for the residue is not defined as a standard amino acid in the graphical program dictionary.)](fig7_p46.gif){kind=link}
{kind=link}
{kind=link}
{kind=link}
![Fig. 12. [46k] The "breathing" motion of NADPH during the MD simulation of wt DHFR complex.](fig12_p4.gif){kind=link}