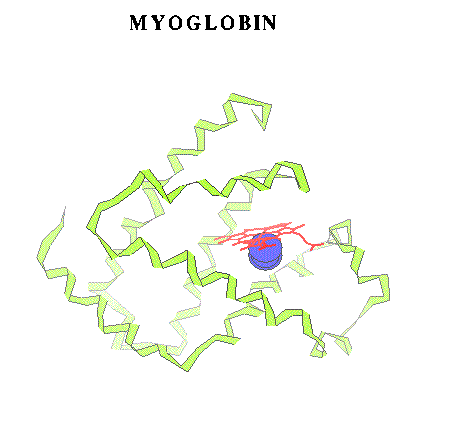
Fig.1 (Schematic view of myoglobin and a diatomic ligand in the distal pocket.)
(b) Department of Computer Methods, N. Copernicus University,
ul. Grudziadzka 7, 87-100 Torun, Poland
A subtle changes of a conformation in the critical heme group modulates the affinity of Hb towards its natural ligand - dioxygene. This ingenious model of protein action, based on earlier works [2], definitely gave a strong impulse for developement of a new field of molecular dynamics (MD) of biomolecules [3]. Computer models of proteins help a lot in relating their structure and activity [4], in indicating paths for the diffusive part of a ligand motion towards the heme reaction centre [5] or even make possible to calculate whole classical reaction paths in heme proteins [6]. All new experimental findings, for example, a recent discovery of resting sites for the CO ligand photodissociated from mioglobin (Mb) [7] (Fig.1), are immediately scrutinized in terms of computer models [8,9].
Heme proteins are especially attractive objects of the theoretical research. The heme unit embedded in a unique polypeptide environment allows for various functions including: the oxygen transport (Hb,Mb), molecular signaling (NOsynthase), redox and CT reactions (cytochromes) [10]. The early successes of MD simulations of proteins were in a large part related to the development of a good forcefield for Mb [11-12]. Such work is continued now (see for example [13]).
Myoglobin - perhaps the most popular heme protein - may be viewed interactively here using coordinates in the pdb format (150K).
However, many crutial features of Mb (or other heme proteins') activity can't be described by classical mechanics. For example, the Fe-ligand bond breaking process requires quantum-mechanical description. The rapid heme doming after the oxygen dissociation in Hb seems to be related to a change in the spin state of the central ion [14]. This is also a quantum effect. Thus, there is a high demand for reasonable molecular models of the heme proteins. Obviously, the Mb itself (see Fig. 1) or even the heme group alone ( Fig. 2) are too large for an efficient quantum treatment in dynamical studies.
Fig.1 (Schematic view of myoglobin and a diatomic ligand in the distal pocket.)
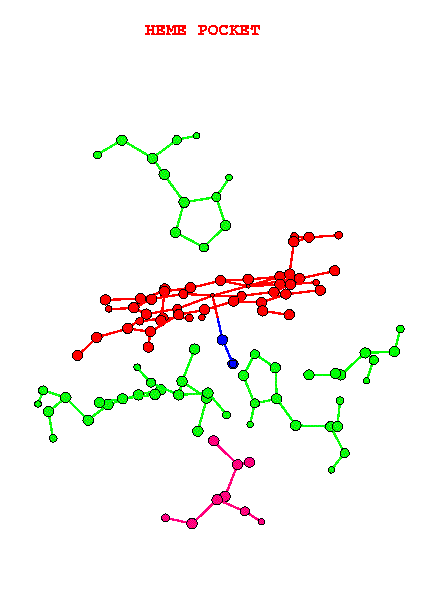
Fig.2 - The heme prosthetic group.
We are not familiar with any calculations which would take into account side chains of the aromatic porphyrine system. Usually simpler molecules are used in quantum calculations. Typical models are presented in Fig.3-6.
Virtually all existing quantum-chemical methods have been used in studies of systems which were suposed to mimic heme reaction centre. In the pre-computer era theoretical studies of the electronic structure of prophyrins the Extended Huckel Method was applied to explain basic features of absorption spectra [15]. For many years the most advanced calculations were GVB results obtained by Goddard and Olafson for a simple square of the four NH2 groups and an ammonia ligand instead of the proximal histidine (Goddard model) (Fig. 5) [16]. It is worthwile to note here a series of papers by M.-M.Rohmer devoted to SCF [17] and even post-HF studies of various models of the heme [18]. Simultanously Kashiwagi et.al. studied, also at the SCF level, electronic and energetic effects of the heme doming [19]. Perhaps the first calculations in the spirit of the DFT for a Mb model had been performed by Case, Huynh and Karplus (x-alpha method)[20]. More elaborated questions regarding a nature of the dissociation of ligands from the heme centre had to be addressed on the semiempirical ZDO level. For example, Loew et.al. calculated electronic spectra of complexes of CO [21], O2 [22] and NO [23] with iron porphyrine and the imidazole as the fifth ligand. Stavrov on the other hand used a ZDO method to explain features of vibrational spectra of heme proteins [24].
A very comprehensive review of calculations for the dioxygen complexes has been recently published by Bytheway and Hall [25]. The same issue of Chem. Rev. contains many intersting papers on heme proteins. A review paper with calculations devoted mainly to the nitric oxide reactions with a typical heme model centre (Fig. 3) was more recently presented by Nowak and Martin [26]. In that study relativistic ECP potentials were used to calculate selected ROHF crossections through doublet potential energy surfaces (PES) for NO reactions with the heme.
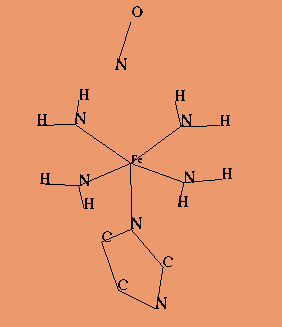
Fig 3. Fig 4.
A typical heme model Imm1NO model
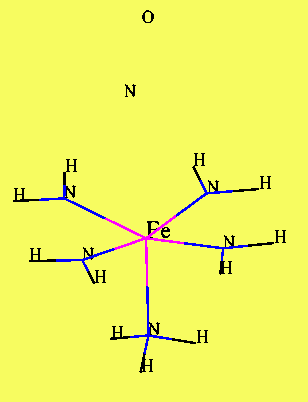
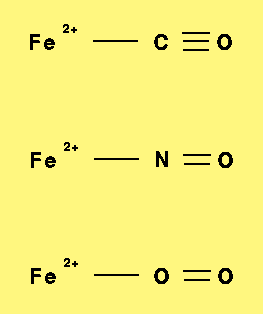
Fig 5. Fig 6.
Goddard model Ionic model
Interesting results on the MP2 level for model system of prophyrin macrocycle simplified to two amidinato ligands have been obtained by Jewsbury et. al. [27]. Using a series of X-ray geometries to model proximal and distal histidnes (imidazoles) these authors noticed that the CO ligand orientation in Mb is largely determined by a special orientation of the proximal histidine. This statement has been recently challenged by results of the DFT calculations of PES for the carbonyl tilting and bending in carbonmonoxyheme [28].
Clearly the HF methods itself, although illuminating as far as basic features of the bonding and electronic strucure in heme proteins are concerned, are not good enough to give a quantitative description of energetics of the dissociation of ligands. Even a correct description of the lowest electronic state in iron porphyrin requires taking into account electron correlation effects. Substantial progress into this direction has been made by Kashiwagi et. al. in their series of CAS SCF calculations for dioxygen complexes of the heme [29]. Very advanced SAC/CI method was used by Nakatsuji et. al. to calculate positions of a few the lowest excited singlet and triplet states of the oxyheme [30]. A good description of the lower absorption spectrum of this molecule was obtained. Moreover a high sensitivity of the position of the lowest triplet state to detailes of the molecular geometry found in this calculation indicates again on a geometrical control of the oxygen affinity of heme.
The only practical approach for using the best quantum methods for studies of biomolecules with transition metal (TM) centers is to select an appropriate mimetic system. A nice example of such an approach is the study by Roos et. al. on electronic spectrum of plastocyanin [31]. The CASSCF/CASPT2 method was used for model complexes.
The electronic structure of TM systems are very difficult to calculate and for a long timescale dynamics only approximate models will be used in the heme protein modelling. Even "tune-ing-up" classical models, such as Landau-Zener [32] or inventing empirical potentials [33] requires some understanding of the simplest reactions of the metal centre with ligands. Such computational studies had been undertaken in the past, but only for the atomic iron interactions with the CO ligand [34,35].
In this presentation some preliminary results of the DFT studies of perhaps the simplest quantum model of the heme reaction centre will be given (sec.4.1). The main goal of this research was to explore the performance of the DFT method in a description of the interaction of the ferrous iron with the typical biological diatomic ligands. The systems choosen (see also Fig.6 ) are small enough that more advanced ab initio method, including CAS SCF/MP2 could be used in the future to collect benchmark information on potential energy surfaces.
The need for a multireference description of the reaction of NO with the heme model system is illustrated in Sec. 4.2 by results of the ROHF calculations of the "binding curves" within the Goddard model.
The ROHF calculations for the systems presented in Fig. 4 and Fig. 5 were performed using US-GAMESS computer code [37]. The relativistic SBKJ potentials were used on all heavy atoms and the standard SBK basis set was empolyed [38]. The geometry of the heme model was taken form [16] and for the imidazole the geometry form [20] was used.
Calculations were performed on the following computers: ymp-el, hp735 (ENSTA-LOA,Palaiseau, FRANCE); Sun UltraSparc e6000, HPC-UMK, SGI O2 - IF UMK, (Torun, Poland).
InsightII software was used for the figures preparation [39].
The geometries of ionic models (Fig. 6) were optimized for different spin states of the systems studied. Results are presented in Tab. 1.
| Ligand LO | State | Energy (h) | rFeL | rLO | FeLO (deg.) |
|---|---|---|---|---|---|
| CO | 1A' | -1376.102 94 | 1.905 | 1.124 | 170.8 |
| 3A' | -1976.153 14 | 2.026 | 1.123 | 167.1 | |
| 5A'' | -1376.200 35 | 2.082 | 1.122 | 165.3 | |
| NO | 2A | -1392.809 69 | 1.793 | 1.111 | 180.0 |
| 4A'' | -1392.849 56 | 1.814 | 1.114 | 180.0 | |
| O2 | 1A | -1413.172 71 | 1.846 | 1.201 | 176.1 |
| 3A | -1413.220 29 | 1.911 | 1.202 | 169.8 | |
| 5A'' | -1413.205 70 | 1.901 | 1.202 | 172.8 |
For the seclected states we have performed calculations of the potential energy curves for the dissociation of the ligand. In each case the Fe-L distance was varied but the equillibrium distance of L=O and bond angles were maintained. As the initial guess molecular orbitals from the previous (shorter Fe-L) convergent calculations were used.
Resulting curves for the FeCO singlet state are presented in Fig. 7.
Fig.7.The potential energy curve for the dissociation of CO.
( postscript version of Fig. 7)
In the case of NO two limiting geometries of Fe-NO unit were studied - the optimized linear and the bent one (Fe-N-O bond angle was 145 deg., N=O bond length was 1.12 Ang.
Fig.8. The potential energy curves for the dissociation of NO:
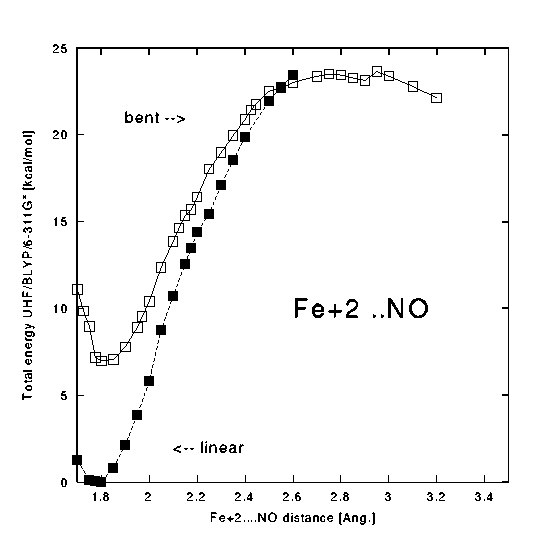
( postscript version of Fig. 8) For the dioxygen ligand a triplet PES was studied:
Fig.9.The potential energy curve for the dissociation of O2.
( postscript version of Fig. 9)
Results of ROHF sample calculations for the modified Goddard model (imidazole instead of NH3) and the simpler one are presented in Fig. 10 and Fig. 11, respectively.
Fig.10.The ROHF potential energy curve for the dissociation of NO.
Doublet state.
( postscript version of Fig. 10)
Fig.11.The ROHF potential energy curve for the dissociation of NO.
Doublet state.
( postscript version of Fig. 11)
For the Fe-NO system the lowest doublet state has been found to be 25.0 kcal/mole above the quartet state. The same tendency for increasing the Fe-NO bond length upon an incerease of the spin is found here (1.793 A - doublet and 1.814 A - quartet). The DFT/BLYP method predicts a linear geometry for this system. The Mulliken charge of iron is lower in Fe-NO than in Fe-CO (1.55 - 1.59).
The lowest spin state for the Fe(+2)-O2 molecule is quintet (see Tab.1). The next triplet state is located 9.2 kcal/mole higher and the singlet state is still 29.9 kcal/mole higher than the triplet. Also this molecule is non-linear in our optimization. In the singlet state the Fe-O2 bond length is substantially shorter than in the other states.
The DFT method with BLYP functional, as was shown by Handy [40], provides a reasonable description of the dissociation process. Thus the dissociation curves were estimated using the UHF formalism for selected states of our ionic models. We found that a general shape of these curves in all three cases is reasonable. However, the "real" meaning of this cross sections, as far as particular spin states are concerned, should be interpreted with a caution, since the observed spin contamination, especially at larger distances is very high. (Note that the expectation value of the S2 operator in DFT may not necessarily be a good observable.)
In all cases studied, at Fe-L distance of 2.6-3.0 Ang. we encountered serious problems with the SCF convergence. Perhaps the difficulty in obtaining of a nice dissociation limit in these simple models is related to the bereakdown of the one-determinant approximation to the calculated states, or to a higher density of states in the inflection region. Other possibility is just an inadequate mesh used in the numerical integration - no allowance has been made in these calculations for the increased size of the system studied.
Despite the lack of real dissociation limits in our DFT calculations, one can estimate that the binding energy in the singlet Fe+2-CO is the largest (>65 kcal/mole). This estimate for Fe+2-NO (doublet) is of 24 kcal/mole and for Fe+2-O2 of about 8 kcal/mole). Again, also these numbers are rather crude estimates of real stabilities of ionic models, since no ZPE correction nor BSSE were taken into account, yet.
The ROHF "binding curves", presented for Goddard model of NO rebinding to the heme (Fig. 11) or for improoved model (Fig. 10), clearly indicate that a single determinant description of this process is inadequate. The curves show only very weak minima (<5 kcal/mole) at rather large (>2A) distances. Since these pilot calculations were performed for rigid geometries, the convergence to the excited state can not be ruled out. On the other hand in the case of Goddard model an intersting "jump" of unpaired electron is observed at 2.5 Ang. (from NO to Fe). It would be very interesting to compare these curves with DFT data for the same system. Such calculations are in progress in our laboratory.
The more advanced model of the heme, studied so far at the ROHF level, doesn't reflect the presence of a rather strong NO-Fe bond observed in nitrosylhemoglobins.
With this presentation we would like to induce a discussion on the best way of a quantum-chemical description of the heme proteins. All comments are very welcome.
This study was performed within the project UMK 389-F. Partial support from the Polish State Commitee for Scientific Research grant no. 6 P04A 032 11, project BiMol (FNP) and INSERM (France) is also acknowledged.
{kind=link}
{kind=link}
{kind=link}
{kind=link}