{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
EMail: wiesiek@phys.uni.torun.pl
Recently synthesized acetoxy derivatives of phenylanthracene are highly fluorescent compounds. They may be used as laser dyes and fluorescent probes of a biomolecular structure [J.R.Heldt, J.Heldt and K.Aleksandrzak, J.Photochem.Photobiol. A:Chem., 81 (1994) 79.]. The rational application of these compounds in spectroscopic studies, e.g. fluorescence anisotropy measurements, requires detailed knowledge of the geometries and the transition moment directions.
This contribution presents the first systematic INDO/S semiempirical study of the singlet absorption spectra of the series of the following molecules:
Spectral studies of large organic molecules have a very long tradition. The unexpected observation of light emitted from irradiated quinine resulted in the discovery of fluorescence by George G. Stokes [1]. A critical analysis of spectra often yields new ideas in physics and chemistry. For example, a brilliant interpretation of spectral studies of organic dyes lead Aleksander Jablonski to the concept of the famous Jablonski diagram [2], now present in many textbooks [3]. Thus, virtually all newly synthesized organic compounds are studied with respect to their spectral properties.
Preliminary studies of the electronic spectra of a group of acetoxy anthracene derivatives which were firstly made by J. Gronowska et. al. [4,5] indicate that they have lasing properties [6]. Recently, a possiblity of a new application of these compounds as fluorescent probes of viscosity [7] was investigated in our laboratory [8]. Fluorescence anisotropy decays were measured in the nanosecond domain using time-dependent spectroscopy for a series of compounds presented in Fig. 1. These data have been the basis for a rotational diffusion model for each compound. Such a model gives a description of the hydrodynamic shape of the molecule. The interpretation of the results of the anisotropy measurement depends to large extent on a knowledge of the electronic transition moment (TM) directions in a probe molecule. [9].
An experimental determination of the TM direction is possible, but this requires a rather tedious procedure [10]. The fluorophores should align well in an ordered environment and such a condition puts a restriction on the molecular structure of the probe. On the other hand, spectroscopically parametrized quantum-chemical (QCH) methods offer a fast and efficient way for the estimation of the TM directions [11]. Moreover, a theoretical approach gives a practical tool for studies of TM directions with respect to geometrical transformations. This feature is particularly useful in studies of flexible molecules that adopt many conformations in solution. Information stemming from QCH calculations is an important part of a general scheme for selecting an adequate rotational diffusion model proposed recently by Szubiakowski et. al. [9]
The goal of the present study was to obtain information on the directions of absorption TMs for a series of acetoxy derivatives of phenylanthracene. TMs of emission are equally important in the modelling of rotational diffusion, but their calculation requires a knowledge of the geometry of the second singlet state. An optimization of the geometry of the excited state of the same symmetry as the ground state is not a trivial task and in this paper we will consider only absorption electronic transitions.
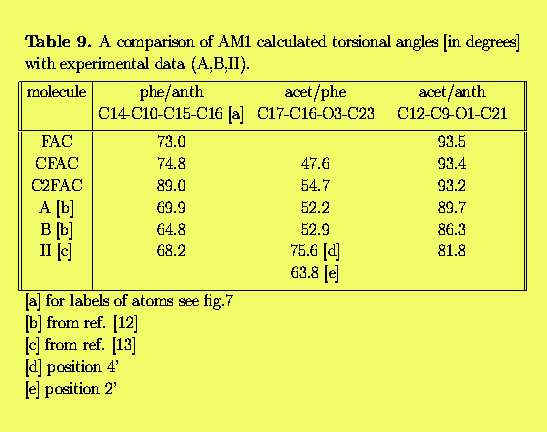
Additionally, we would like to get some insight into the rigidity of the 9-phenyl-anthracene derivatives - mainly with respect to conformations of the acetoxy group in solution. The crystallographic structures of similar derivatives were determined by Roszak et.al. [12]. In that paper the structure of 9-acetoxy-10-(2-acetoxy- bromophenyl)anthracene is reported while in [13] the 9-acetoxy-10-(2,4-diacetoxyphenyl)anthracene is described. Thus, we would like to check to what extent a standard QCH method like the Austin Model 1 (AM1) [14] is able to reproduce the x-ray geometry and predict reasonable geometries of the other compounds from this group.
It is worthwhile to note that results presented here have also a biophysical aspect since the anthracene derivatives are often used as probes of the dynamics of biological membranes [3,15].
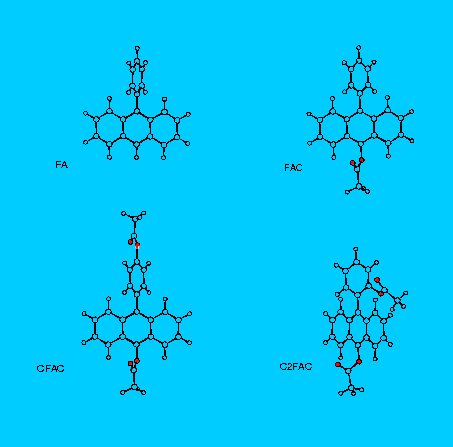
For this study the following compounds were selected:
(interactive molecules in pdb format, use for example RasMol2.5 to see structures)
The size of the molecules studied here puts severe restrictions on the level of the method to be used in calculations of TMs and geometries. The only practical approach is a semiempirical one. For calculation of absorption spectra and TM directions a modified INDO/S method was used. The all-valence INDO/spd algorithm (Ghost and Rydberg INDO), developed by J.Lipinski [16] has been very useful in interpretations of the electronic absorption spectra of large molecules (see for example [17]) and transition moment directions [11]. In calculations of excited state wavefunctions 60 lowest singly excited configurations were included in the CI procedure. The parametr k, scaling ß integrals, was set to be 1.0.
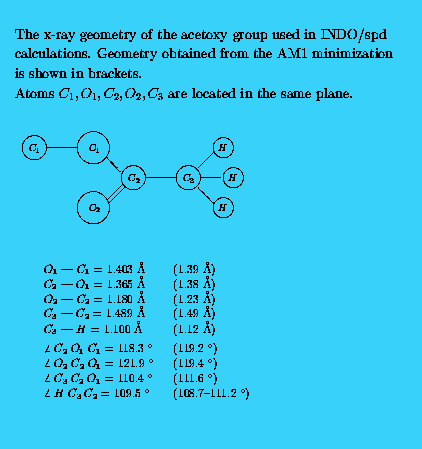
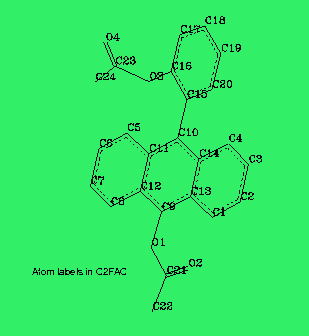
All geometries for INDO/spd calculations were based on x-ray data obtained from [12], however, a small deviation from planarity of the anthracene group was discarded. A typical geometry of the acetoxy group is presented in Fig. 2. The anthracene rings were always located in the XY plane, the X axis was oriented along the short axis of the anthracene. The calculations of spectra were performed on a PC386/387 computer.
The geometry of the compounds studied was also optimized with the AM1 method [14]. Initial structures were built using InsightII v. 2.3.5 (Biosym Technologies) program and calculations were performed with MOPAC 6.0 code [18] implemented in InsightII. Geometries were optimized without constraints, using PRECISE option. That part of calculations was performed on a SGI Indy workstation.
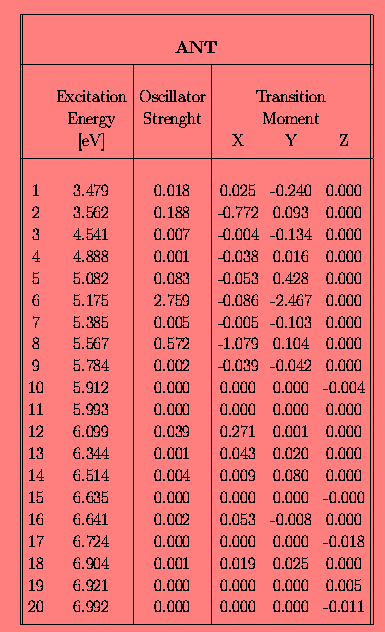
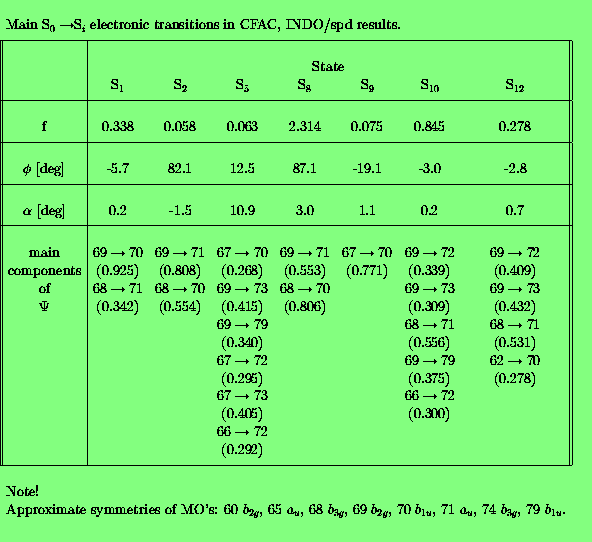
Since our goal was to study electronic TM directions in anthracene derivatives we have firstly checked how the INDO/spd method reproduces an absorption spectrum of a bare anthracene. The results of calculations for the 20 lowest singlet states are presented in Tab. 1.
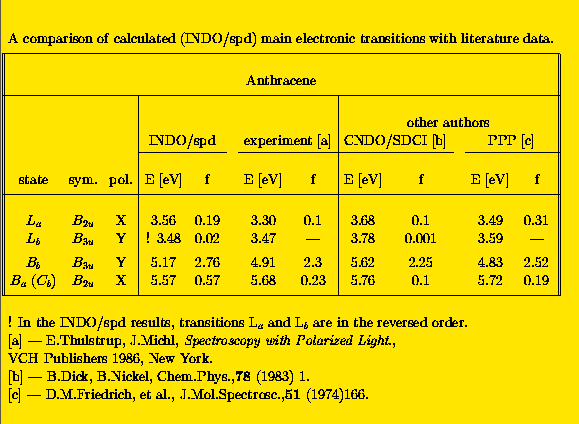
Note, that in this calculation no Dh2 symmetry was imposed since the geometry of anthracene was "extracted" from x-ray data [13]. The calculated energies of electronic transitions, TM directions and oscillator strengths are in a good agreement with the experimental ones. A comparison of a few spectroscopically important excited states with the literature data is presented in Tab. 2.
Calculated transition energies are about 0.2 eV higher than experimental ones observed in a cyclohexane solution. The INDO/spd method correctly describes near degeneracy of B2u and B3u states corresponding to the first absorption band of anthracene, however, their order is different from that predicted by the other methods. Perhaps this inconsistency may be attributed to the lack of symmetry of the anthracene moiety. It is worthwile to note that the experimental determination of the position of this very weak 1B2u+ state was a subject of a long controversy. A detailed discussion of the electronic spectra of anthracene may be found in [10].
Both polarization experiments [10] and our calculations indicate that the lowest absorption band of anthracene consists of two perpendicularly polarized transitions: a strong one with a short axis polarization, and very weak one having a long axis polarization. In the second band (Bb) one very strong transition dominates. It is long axis polarized. The third band (Ba), according to the present INDO/spd results, is short axis polarized and there are no other highly excited states with the perpendicular orientation of TMs.
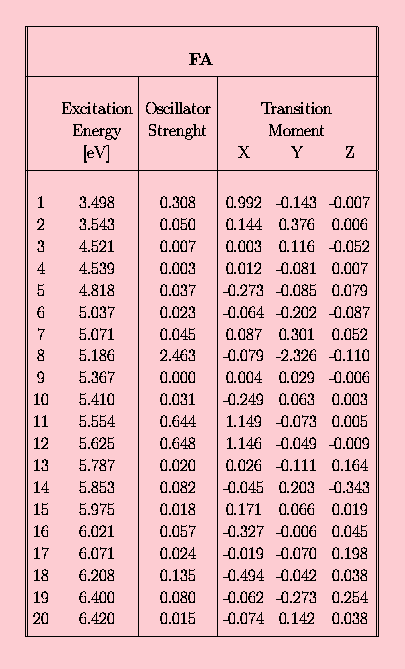
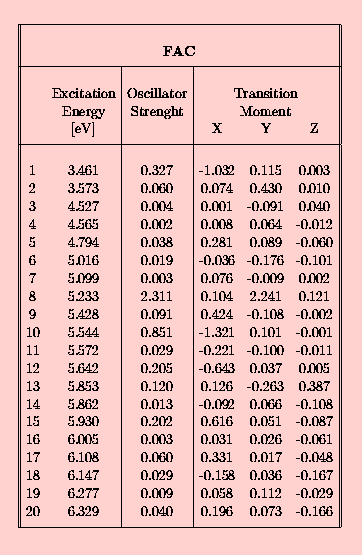
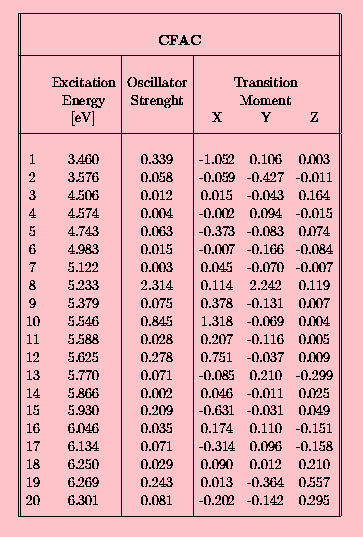
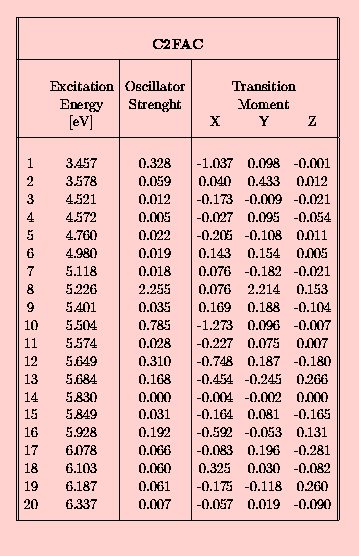
In the following set of tables the INDO/spd results are presented for the 10-phenylanthracene (FA) in the Tab. 3., and for acetoxy derivatives of FA, namely FAC in Tab. 4., CFAC - Tab. 5. and C2FAC in Tab. 6..
The calculated INDO/spd electronic spectra of FA and it's acetoxy derivatives are similar. In all compounds the lowest singlet state is of B2u (La) approximate symmetry. The addition of the phenyl group to the anthracene rings results in an increase of the oscillator strength from 0.188 to 0.308 (FA case). The presence of the acetoxy substituent in the anthracene ring causes a further small increase of this value, for example f(S0-->S1) in FAC is 0.327. The addition of the acetoxy group to the phenyl ring does not affect the oscillator strength of the first transition. Transition energies to the S1 state in the phenylanthracene compounds are lower than in the anthracene by about 0.05 eV (FA) and 0.1 eV (FAC,CFAC,C2FAC). The 0-0 component of the first absorption band of FAC is redshifted by 1080 cm-1 with respect to ANT in a dioxane solution [21]. The calculated red-shift of 800 cm-1 is in an agreement with this observation. The INDO/spd transition energies of acetoxy derivatives are about 0.3 eV higher than 0-0 transitions observed in dioxane. For example, in FAC the calculated value is 27 900 cm-1 and the observed absorption band is located at 25 350 cm-1 [21]. Such a difference is typical and is clearly connected with the absence of a polarizable medium in the QCH calculations.
In all phenylanthracene compounds studied here the weak B3u (Lb) state is located above the B2u (La) state. However, the B3u-B2u gap increases substantially from 0.05 eV (FA) to 0.1 eV after an attachement of an acetoxy group to the anthracene subsystem.
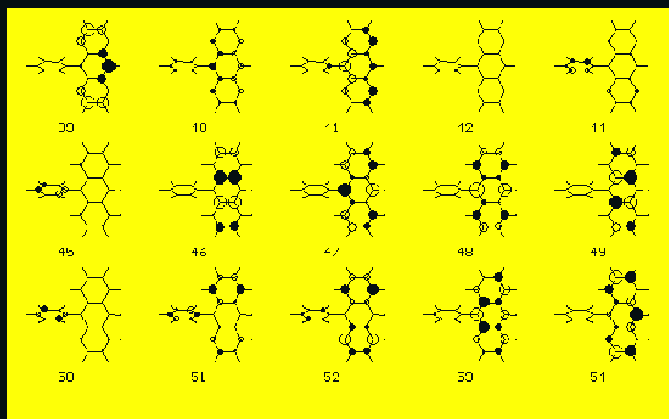
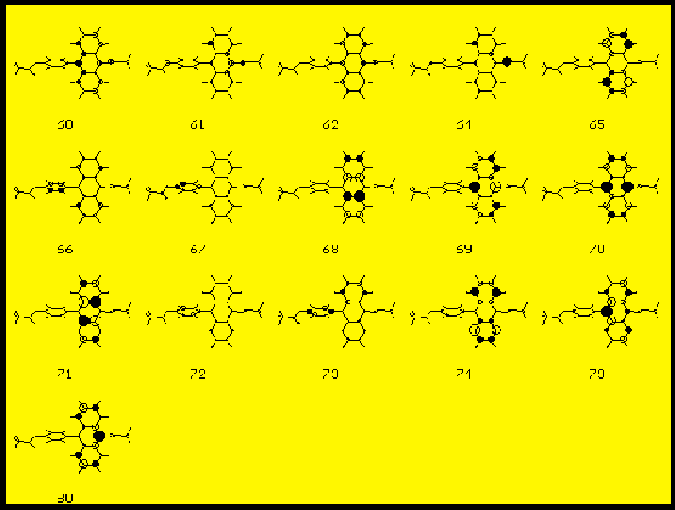
The compositions of the main electronic transitions in FA are presented in Tab. 7. and the corresponding molecular orbitals are shown in Fig3. The same data for the CFAC derivative are displayed in Tab.8 and in Fig. 4. In all compounds studied in the first strong transition (S1 state) the HOMO --> LUMO excitation dominates. In the second absorption band region (state S8) we also observe almost an identical composition of the CI vector for all derivatives. Main contributions come from HOMO-->LUMO+1 and HOMO-1 --->LUMO one-electron excitations. In low lying singlet states with f>0.05 we do not observe any contributions to pi MOs from acetoxy groups. All important transitions are localized on anthracene.
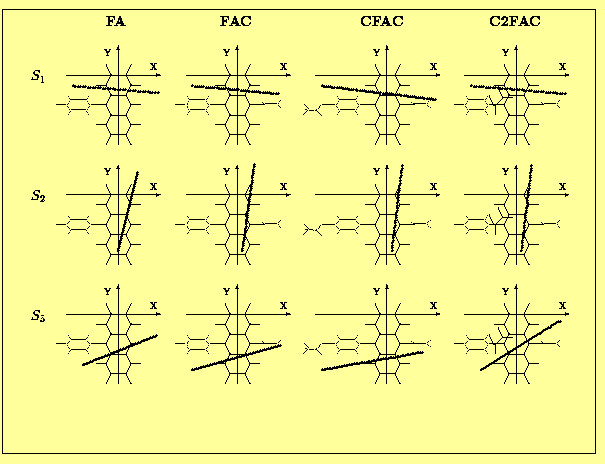
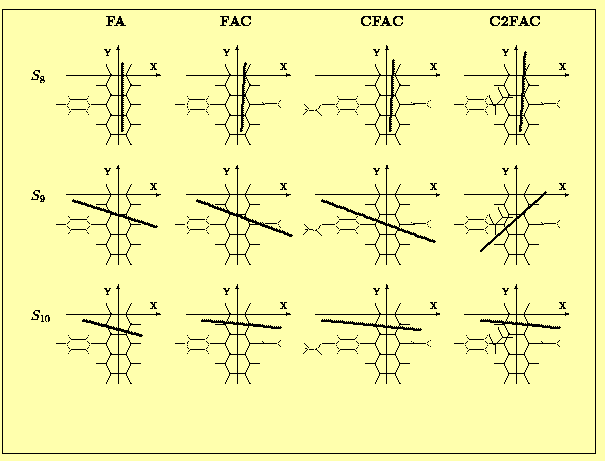
The angles between the calculated TM directions and the OX axis (the upper row) and the XY plane (the lower row) are presented in Tab.7 and Tab. 8. In almost all spectrally interesting states (energy <5.5 eV , f>0.05) the transition moments are located in the anthracene plane. The only exception is the S5 state of CFAC where the TM is tilted by 10 deg. with respect to this plane. In this particular case we observe a substantial contribution from transitions localized on the phenyl ring of CFAC (see Tab.8 and Fig. 4 ) to the CI wavefunction of the S5 state.
The most interesting, from the point of view of experimentalists are data - directions of the spectroscopically relevant TMs, which are shown schematically in Fig. 5a (states #1,2 and 5) and Fig. 5b (states #8,9 and 10). One of the most important results of the present calculations is the observation that the polarizations of the S1,S2,S5 and S8 states do not depend on the presence or position of the acetoxy group. The S9 state of C2FAC exhibits a different polarization than that observed in the other derivatives, but one should note that the order of the higher lying states (energy>5.5 eV) does depend on the phenylanthracene substituents. The proximity of singlet exited states with a rather large ocillator strengths and perpendicular TM directions is the most probable explanation of a large deficit in limiting emission anisotropy value observed in these compounds [8].
The INDO/S data shown in the previous section were obtained with geometries determined by solid state x-ray experiments. Since the compounds studied here have several nearly planar fragments linked via a single bond, they may perhaps adopt various conformations in solution. For example, fluorescence studies of FA in 2-methylbutane performed by Wortmann et. al. [19]. indicate clearly that this molecule exhibits large amplitude motions in room temperature solutions. The AM1 torsional potential calculated by these authors is quite flat in a broad region of phenyl-anthracene torsional angles fi. In the range of fi=(60, 120) deg. the potential energy of FA changes by less than 100 cm-1 (0.3 kcal/mol). The AM1 potential is in very good agreement with the experimental one obtained from a molecular beam experiment by Barbara et.al [20].
In order to get some insight into the possible hydrodynamic shapes of the probes studied and to check the performance of the standard AM1 method [14]. in acetoxy derivatives geometry predictions we have performed a full AM1 geometry optimization of this series of compounds.
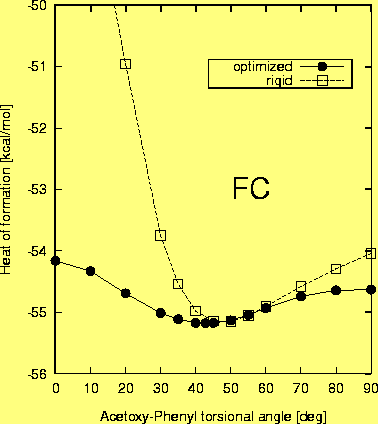
Firstly, however, a rough estimate of the flexibility of the torsion of the acetoxy group around ether oxygene-phenyl carbone single bond has been made. The torsional potential for a rigid rotation of this group, calculated for acetoxybenzene is presented in Fig. 6. The minimum energy conformation was obtained for a torsional angle of 47 deg., but again, this potential is quite flat. The barrier to rotation at 90 deg. is about 1 kcal/mole and full optimization decreases this value further to 0.55 kcal/mole, as indicated by the AM1 potential curve calculated with relaxation of all remaining internal coordinates Fig. 6. The barrier of 1.02 kcal/mole obtained at 0 deg. is twice as big, but it is still relatively low. Thus, we conclude, that in a room temperature solution the acetoxy group may adopt a variety of conformations having perhaps a torsional angle in the range of fi from 30 deg. to 150 deg. with respect to the phenyl plane.
Results of our AM1 calculations are presented in two sets of data: a series of the best geometries of each compound in the pdb format which may be viewed interactively using the Rasmol2.5 program :
and a series of corresponding MOPAC 6.0 archive files, which may be easily imported to the XMOL code:
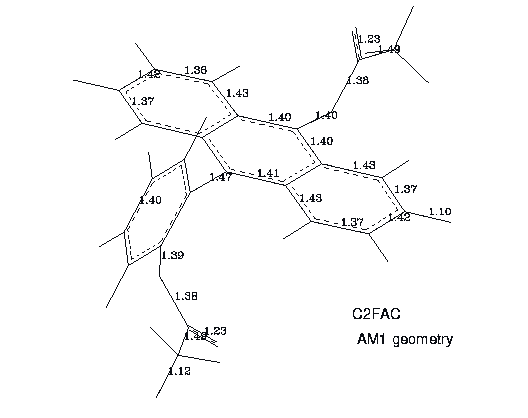
Details of geometries may thus be studied by interested readers equipped with proper software.The AM1 optimized acetoxy groups are planar in all positions studied. The bond lengths and bond angles practically do not depend on the position of the group in the FA backbone. The same feature is observed in crystal structures. The calculated bond lengths are in good agreement with the averaged x-ray data (see Fig. 2). The only exception is C=O carbonyl bond which has a value of 1.23 Ang. in AM1 results but varies from 1.177 Ang. to 1.193 Ang. in crystal stuctures. The bond angle (O1-C2-C3) of 111.6 deg. calculated with AM1 is close to a low experimental value of 110.4 deg. but the other bond angles are not reproduced so well (see Fig. 2).
We have found that in FAC, CFAC and C2FAC the O1 (ether oxygen) atom is located slightly out of the anthracene plane. The same distortion is observed in crystal structures of other acetoxyanthracenes [12,13]. A comparison of the calculated torsional angles between planar fragments with the x-ray data for related structures is presented Tab.9. Since torsional potentials phe/anth and acet/phe are very flat the agreement between theoretical and experimental values of angles is quite good. Interestingly, the AM1 method correctly gives a small increase of 1.1-2.0 deg. of the bond angle at the position 9 of the middle anthracene ring where the acetoxy substituent is attached. In experiment this angle was found to be 3 deg.larger than that in anthracene.
Emission anisotropy experiments indicate, that the C2FAC molecule exhibits in a paraffin solution a different hydrodynamic shape than the other derivatives [8]. The similarity of the TM moment directions of all the derivatives suggests that this observation has to be linked to a specific orientation of the phenyl substituent having the acetoxy group in position 2':
Figure 7. A structure (AM1) of C2FAC molecule.
The selected bond lengths of C2FAC, representative to the other derivatives, are presented in Fig. 8
Figure 8. Selected bond lengths of C2FAC conformer. The AM1 data.
In contrast to the other molecules from the group studied the rotation of the phenyl ring around the phenyl- anthracene bond in C2FAC is severly hindered. Thus, in this molecule an approximate cylindrical symmetry along the phenyl-anthracene axis is lost, and it's diffusion tensor does not coincide with the molecular frame.
The acetoxy groups generate a local dipole moment of about 1.5 Debye (FAC). In the derivative CFAC contributions of the two dipoles cancel out and the electric dipole moment has a value of 0.7 Debye. In the C2FAC molecule the highest dipole moment of 2.6 D was calculated. A charge distribution obtained with the AM1 method for C2FAC is presented in Figure 9:
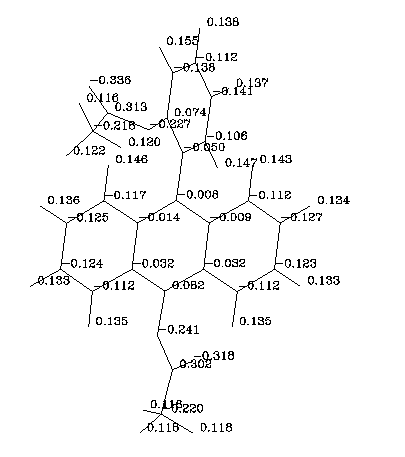
Figure 9. Partial charges in C2FAC molecule. The AM1 data.
These data indicate that in polar protic solvents (for example in glycerole) acetoxy groups should make numerous hydrogen bonds with solvent molecules. We expect therefore that rotational diffusion times will be much larger than observed in paraffin solutions. The experiments checking this prediction are under way in our laboratory.
All derivatives studied are flexible molecules. They may adopt a range of conformations in solution, but the C2FAC molecule has a rather rigid orientation of the phenyl part with respect to the anthracene subsystem. The AM1 geometry is in reasonable agreement with the published x-ray data for related compounds but the calculated torsional angles for isolated molecules are different from those observed in crystals. The AM1 method correctly predicts the planarity of the acetoxy group but the C=O double bond is much longer than experimental one. In our opinion the combined AM1 and INDO/spd calculations give a good estimation of the transition moment directions of flexible fluorescent probes and should be helpful in the interpretation of further fluorescence polarization experiments.
A support from the Polish State Commitee for Scientific Research grant no. 2 P302 205 04, project BiMol and UMK grant no. 382-F is acknowledged.